Nearly 3 percent of the world’s population has some form of psoriasis—that’s over 125 million people. Of those, an estimated 7.5 million are Americans, according to the National Psoriasis Foundation (NPF), making it the most common autoimmune disease in the country.
Although this skin disease is prevalent, many people are still unaware of its impact. Unfortunately, there are many misconceptions about the disease; for example, that it is contagious.
What is Psoriasis?
Psoriasis isn’t just a skin disease; it is actually am autoimmune condition that has the potential to cause widespread systemic effects. These widespread systemic effects are most commonly described as effects on the skin, joints and heart. There are different forms of Psoriasis and some are more common than others.
Psoriasis is a chronic autoimmune disease that causes skin cells to multiply up to 10 times faster than normal. This makes the skin build up into bumpy red patches covered with white scales that can grow anywhere, but typically appear on the scalp, elbows, knees, and lower back. Psoriasis is not contagious nor is it caused or worsened by poor personal hygiene. Psoriasis may be inherited and can range from a very mild, hardly noticeable rash to a severe eruption that covers large areas of the body. Affiliated Dermatology’s Dr. Andrew Newman shares some facts about psoriasis:
“Although psoriasis is typically thought to be a condition that only affects the skin, it affects the ENTIRE body. In fact, joint disease, heart disease, and depression are common features in psoriasis. It’s caused by many factors including genetic predisposition, certain medications, and some infections such as strep throat. People with psoriasis most often are regularly taken care of by a dermatologist.”
In some patients, psoriasis causes nail changes and joint pain (psoriatic arthritis). The first episode usually strikes between the ages of 15 and 35. This chronic condition will then cycle through flare-ups and remissions throughout the rest of the patient’s life.
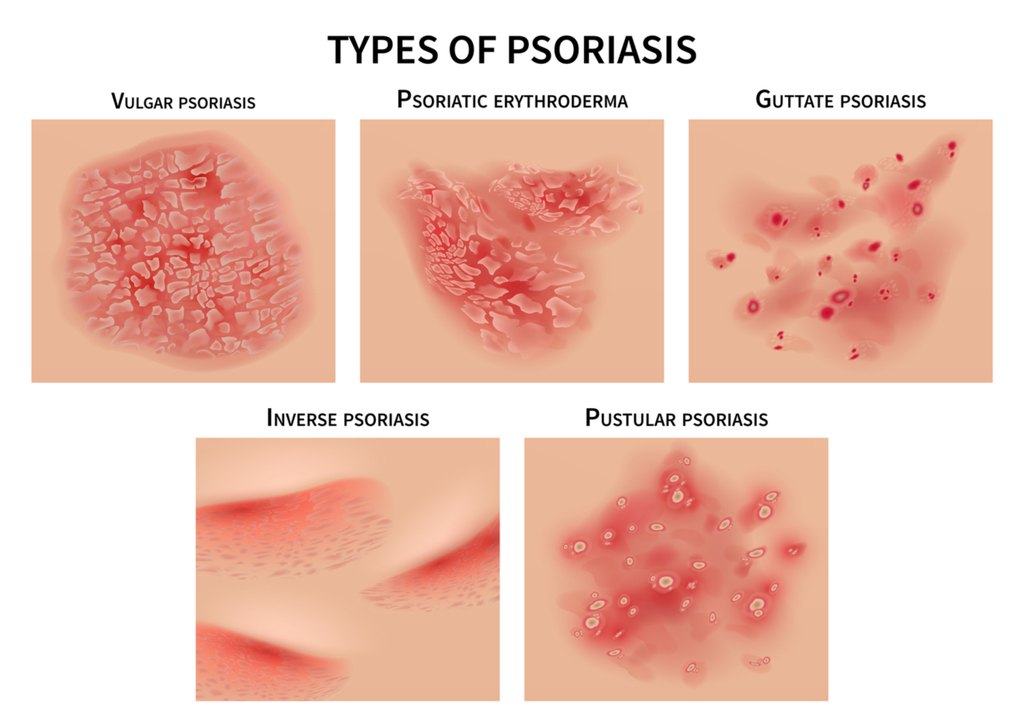
What are the different forms of Psoriasis?
The most common form of the disease, plaque psoriasis, appears as raised, red patches covered with an accumulation of white dead skin cells. Other areas affected by the different types of psoriasis include the face, skin folds, hands, feet, genitals, and nails.
Most individuals will be afflicted with one form of Psoriasis at a time, there are known treatments for Psoriasis, but currently there is no known cure. Occasionally, when one form of Psoriasis clears up and symptoms reside, another form may appear due to exposure to a trigger. Triggers include but are not limited to; skin injury, stress, certain medications, infections, weather, diet and allergies.
1. Plaque Psoriasis – Plaque Psoriasis is the most common type of Psoriasis; it can also be called ‘Psoriasis Vulgaris’. This type of Psoriasis appears as red, inflamed patches of skin covered with a white or silvery buildup of dead skin cells (known as plaque). It can cause the skin to feel painful to the touch and itchy and typically effects the knees, elbows, scalp or lower back, however … it can occur anywhere on the body.
2. Inverse Psoriasis – Inverse psoriasis makes bright red, shiny lesions that appear in skin folds, such as the armpits, groin, and under the breasts.
Inverse Psoriasis appears as areas od shiny, red and inflamed skin. This type of Psoriasis is typically located in the folds of the body; under the arm pits or breasts, behind the knees, around the groin or even the skin folds that surround the genitals.
3. Guttate Psoriasis – Guttate psoriasis often starts in childhood or young adulthood, causes small, red spots, mainly on the torso and limbs. Triggers may be respiratory infections, strep throat, tonsillitis, stress, injury to the skin, and taking antimalarial and beta-blocker medications.
Guttate Psoriasis will more commonly present in childhood or amongst young adults. Symptoms appear as small pinkish-red spots or lesions, typically on the arms, legs and torso.
4. Erythrodermic Psoriasis – Erythrodermic psoriasis causes fiery redness of the skin and shedding of scales in sheets. It’s triggered by severe sunburn, infections, certain medications, and stopping some kinds of psoriasis treatment. It needs to be treated immediately because it can lead to severe illness.
This is one of the least common types of Psoriasis but it one of the most serious. More severe symptoms include severe burning, itching and peeling of the skin, changes in body temperature and a faster heart rate. If you believe you are suffering from this type of Psoriasis, see your doctor immediately, it can cause severe illness.
5. Pustular Psoriasis – Pustular psoriasis causes red and scaly skin with tiny pustules on the palms of the hands and soles of the feet. Pustular Psoriasis is another typically uncommon type of Psoriasis that mainly appears in older adults. Symptoms include pus-filled bumps, known as pustules, the surrounding skin can appear red and inflamed, oftentimes looking infectious (however, it is not). This type of Psoriasis can appear mainly on the hands and feet but can appear on other parts of the body as well. Symptoms of Pustular Psoriasis can include; nausea, fever, chills, muscle weakness, and rapid heart rate.
6. Psoriatic Arthritis – Psoriatic Arthritis is a variant of the condition where the individual has both arthritis (joint inflammation) and psoriasis. Typically, this condition appears years after the onset of Psoriasis symptoms. Symptoms can include; warm or discolored joints, swelling of the joints – fingers and toes, and stiff, painful joints that are worse after rest or in the mornings.
7. Nail Psoriasis – Nail Psoriasis is another variant of the condition and it more commonly affects those who are afflicted by Psoriatic Arthritis. Symptoms can include; painful, tender nails, color changes to the nails, a white chalk-like material under your nails, pitting of your nails, separation of the nail from the nail bed.
8. Scalp psoriasis can cause dandruff-like itching and flaking. Psoriasis happens when the immune system triggers too many skin cells to grow on various parts of the body. That can include your scalp. People with psoriasis may be more likely to get dandruff, but psoriasis is not dandruff.
Living with Psoriasis can affect your quality of life; however, certain treatments are available. You can work with your doctor to develop a plan of care and guide you in figuring out what your environmental triggers are or other lifestyle factors. Triggers and lifestyle factors could be the culprit behind flare-ups.
What causes Psoriasis?
Although psoriasis appears on the skin, it is an immune system disease that is not caused or worsened by poor personal hygiene. People with the disease have a genetic tendency to develop it. There are certain things that can trigger flare-ups including skin injury, stress, hormonal changes, infection, and medications. Most people with the disease experience cycles of clear skin and outbreaks. Dr. Dustin Mullens of Affiliated Dermatology spoke on how Psoriasis starts:
“The nervous system and stress affect a multitude of skin conditions in humans. There are many types of cells in the skin affected such as immune cells and endothelial cells, both can be regulated by neuropeptides and neurotransmitters, which are chemicals released by the skin’s nerve endings. Stress can result in the skin’s nerve endings releasing an increased level of these chemicals and when this occurs, it can lead to inflammation of the skin. This is why people often experience a flare-up of their inflammatory skin conditions such as psoriasis during times of stress.”Things that can trigger an outbreak of psoriasis include:
- Cuts, scrapes, or surgery
- Emotional stress
- Strep infections
- Medications, including
- Blood pressure medications (like beta-blockers)
- Hydroxychloroquine, antimalarial medication
Symptoms of Psoriasis
The truth is that there are many people with psoriasis who don’t even know they have it! Skin rashes are not uncommon so dermatologists need to rule out a list of other possible causes like an allergy to food/medication and viruses. Careful visual inspection is needed for diagnosing psoriasis, but sometimes there is a need for a skin biopsy.
Is infection a possibility? Infections are actually quite rare due to the fact that psoriasis itself is due to an overactive immune system. That being said, repeated scratching and excoriation can disrupt the skin barrier and facilitate bacterial invasion and is thus strongly discouraged. All patients with psoriasis should be seen at the very least annually by a dermatologist and when treatment and medications are ineffective at controlling disease severity and flares. Patients requiring systemic treatment should be seen every 3 months for check-ups while on these more sophisticated/complex medications.
How to Treat Psoriasis?
There’s currently no cure for this chronic autoimmune condition, but caring for psoriasis can slow down the growth of skin cells and relieve pain, itching, and discomfort. Treatment of psoriasis depends on a patient’s overall health, presence of joint pain, and severity of skin involvement. When asked about treatment for psoriasis, Dr. Newman shares,
“The type of treatment used depends on the total body surface area involved and severity, etc. In mild psoriasis, I think natural medicines work well. Some people find benefit from taking the natural anti-inflammatories quercetin and curcumin. Additionally, they may find that applying aloe vera gel to the skin does wonders. Lastly, sunlight also helps with mild psoriasis. That’s right, the UV rays of the sun decrease the skin inflammation in psoriasis! In fact, this explains why my colleagues and I see less psoriasis where we practice in the sun-rich Phoenix, Arizona, compared to areas like the midwest.”
In mild cases, topical corticosteroids and medications are prescribed. Psoriasis is not curable, but it is controllable. No single approach works for everyone. Therapy is individually tailored and based on your health, goals, and a careful assessment of potential risks and benefits of treatment. Treatments can be divided into four main types:
- Topical treatments
- Light therapy
- Systemic medications
- Biologics
Dr. Newman goes on to say, “For more serious psoriasis, it will be almost impossible to successfully manage the disease without sophisticated prescription medicines. Usually, this will entail potent topical corticosteroids and/or certain oral or injectable medicines that help regulate the body’s immune system (which has gone haywire in psoriasis). Importantly, if you have psoriasis (mild or severe), you should discuss the use of both natural and prescription medicines with your primary care doctor and your dermatologist.”
Find Relief for Psoriasis
The best treatment varies by individual, taking into consideration the type of psoriasis you have, where it is on your body and the possible side effects of medications. Another AffDerm dermatologist, Dr. Mitchell Manway, gave us some extra tips on what to do when you have psoriasis.
Moisturizers: which kind are the best? “In general, the thicker or greasier the moisturizer, the better. Creams and ointments that come in a tub or jar are more effective at restoring the skin barrier than lotions or products that come in pump-dispensers. Products containing petrolatum or ceramides can be particularly effective or preferred,” says Dr. Manway.
Scale softening products? What ingredients work best? Dr. Manway advises, “Products that contain lactic acid (Amlactin/Lac Hydrin), salicylic acid (Salex), or urea are more effective at removing scale and improving skin texture.”
Cold showers/cold packs or warm baths/heating pads? According to Dr. Manway, “Ice-packs and heat may be effective at treating symptoms of itch by distracting nerve receptors, but I would avoid exposure to showers or bathing as this may promote further water-loss and drying of the skin.”
Stress relief options like meditation, acupuncture, etc? “Studies directly involving acupuncture and treatment of psoriasis are still inconclusive, with some proposing benefit and others with no significant results. However, anything that can promote stress relief may be helpful at preventing and controlling flare-ups as stress can be a major contributor for worsening of the disease,” said Dr. Manway
Exercise? “Daily or weekly exercise can stimulate and regulate the immune system and decrease stress levels, and thus is an important part of disease management.”
Over-the-counter remedies like calamine lotion? “In my experience calamine lotion is not very effective at reducing itch or pain. Topical preparations that contain pramoxine (Sarna Sensitive) or menthol (Sarna) are preferred. Surprisingly, brief periods of exposure to sunlight and UV rays can also benefit psoriasis, but limited exposure should be stressed due to the increased risk of skin cancer associated with chronic UVA and UVB damage,” said Dr. Manway.
Prescription medications? Dr. Manway agrees, “Rx medications are by far the most effective topical treatment approach available and help to decrease inflammation at the site of disease. Potent topical steroids such as clobetasol or betamethasone are the most common medications prescribed, but other mechanisms such as vitamin D analogues and calcineurin inhibitors can provide significant and adjunct benefits towards the reduction of psoriatic plaques with less risk of long-term local side-effects. When local disease can not be maintained on topical medications or development of psoriatic arthritis is present, systemic oral medications or biologic therapy/injections are necessary.”
When should you see your dermatologist for psoriasis? Look out for any suspicious changes such as lesions that show signs of persistent flaking, scaling, roughness, redness, scabbing, bleeding, or otherwise non-healing areas. These symptoms are uncomfortable and could be an indication of something more serious.
Genetics of Generalized Pustular Psoriasis: Current Understanding and Implications for Future Therapeutics
1. Introduction
2. Genetics of Pustular Psoriasis

2.1. IL36RN
2.2. CARD14
2.3. AP1S3
2.4. MPO
2.5. SERPINA1, SERPINA3
2.6. BTN3A3
2.7. TGFBR2
3. Current and Potential Therapeutic Agents Targeting Immune Mediators in Generalized Pustular Psoriasis
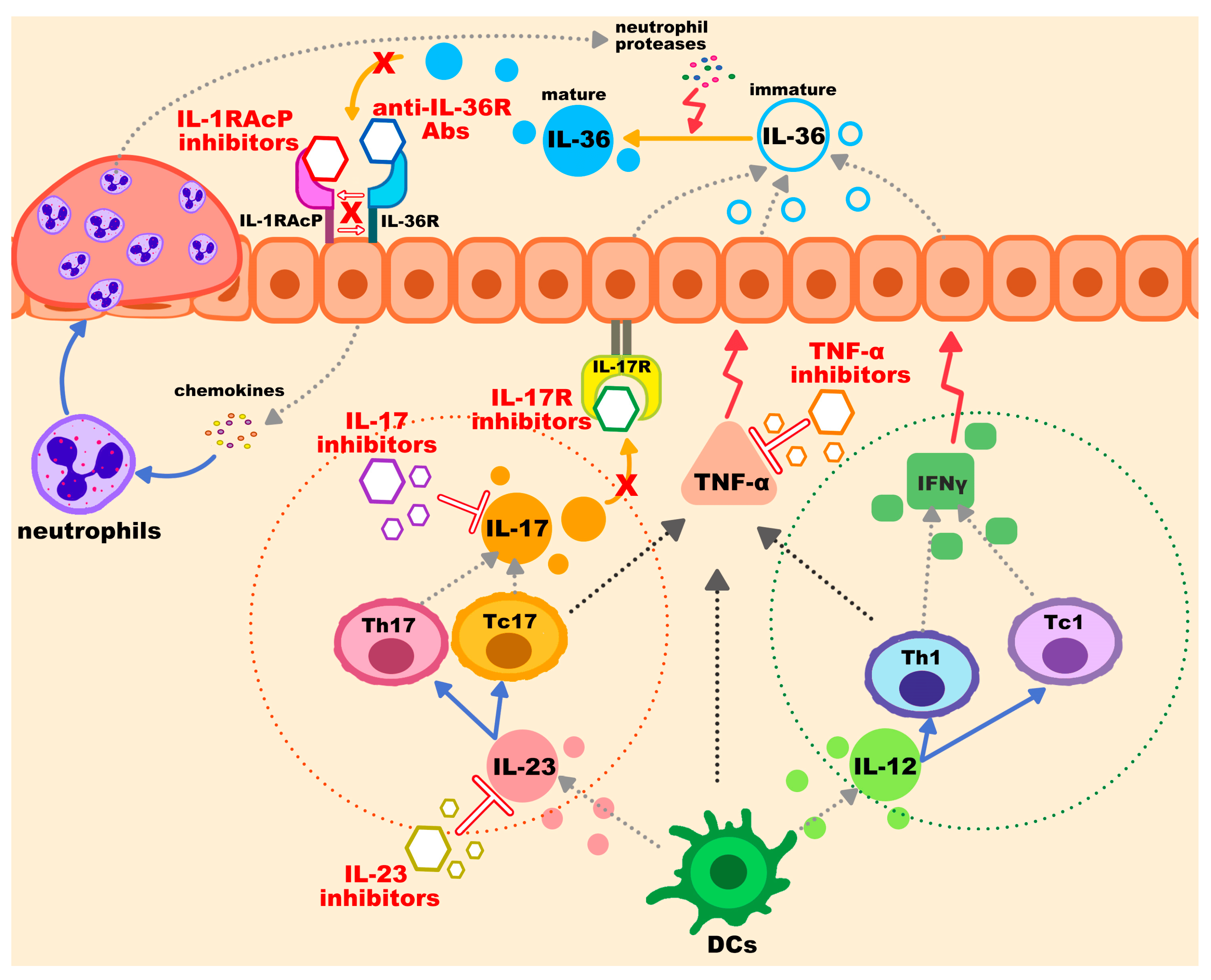
3.1. IL-36 Pathway Inhibitors
3.2. IL-1RAcP
3.3. TNF-α Inhibitors
3.4. IL-17 Inhibitors
3.5. IL-23 Inhibitors
3.6. Additional Biological Therapy and Non-Biologic Options
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffiths, C.E.M.; Armstrong, A.W.; Gudjonsson, J.E.; Barker, J. Psoriasis. Lancet 2021, 397, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Navarini, A.A.; Burden, A.D.; Capon, F.; Mrowietz, U.; Puig, L.; Köks, S.; Kingo, K.; Smith, C.; Barker, J.N. European consensus statement on phenotypes of pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1792–1799. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-C.; Hsu, L.-A.; Huang, Y.-H. Alcohol consumption, aldehyde dehydrogenase 2 gene rs671 polymorphism, and psoriasis in Taiwan. Dermatol. Sin. 2022, 40, 108–113. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Lee, C.-W.; Li, Y.-A.; Chen, T.-H.; Yu, H.-S. Prenatal infection predisposes offspring to enhanced susceptibility to imiquimod-mediated psoriasiform dermatitis in mice. Dermatol. Sin. 2022, 40, 14–19. [Google Scholar] [CrossRef]
- Yu, S.; Tsao, Y.-H.; Tu, H.-P.; Lan, C.-C. Drug survival of biologic agents in patients with psoriatic arthritis from a medical center in southern Taiwan. Dermatol. Sin. 2022, 40, 20–27. [Google Scholar] [CrossRef]
- Mirza, H.A.; Badri, T.; Kwan, E. Generalized Pustular Psoriasis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Twelves, S.; Mostafa, A.; Dand, N.; Burri, E.; Farkas, K.; Wilson, R.; Cooper, H.L.; Irvine, A.D.; Oon, H.H.; Kingo, K.; et al. Clinical and genetic differences between pustular psoriasis subtypes. J. Allergy Clin. Immunol. 2019, 143, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases. J. Allergy Clin. Immunol. 2017, 140, 1545–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, M. Autoinflammatory keratinization diseases: The concept, diseases involved, and pathogeneses. Dermatol. Sin. 2022, 40, 197–203. [Google Scholar] [CrossRef]
- Akiyama, M. Pustular psoriasis as an autoinflammatory keratinization disease (AiKD): Genetic predisposing factors and promising therapeutic targets. J. Dermatol. Sci. 2022, 105, 11–17. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Simpson, M.A.; Pink, A.E.; Di Meglio, P.; Smith, C.H.; Pullabhatla, V.; Knight, J.; Spain, S.L.; Nestle, F.O.; Burden, A.D.; et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011, 89, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.-Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36–Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Luo, Q.; Cheng, Y.; Wen, X.; Liu, J. An update on genetic basis of generalized pustular psoriasis (Review). Int. J. Mol. Med. 2021, 47, 118. [Google Scholar] [CrossRef] [PubMed]
- Setta-Kaffetzi, N.; Simpson, M.A.; Navarini, A.A.; Patel, V.M.; Lu, H.C.; Allen, M.H.; Duckworth, M.; Bachelez, H.; Burden, A.D.; Choon, S.E.; et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 2014, 94, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahil, S.K.; Twelves, S.; Farkas, K.; Setta-Kaffetzi, N.; Burden, A.D.; Gach, J.E.; Irvine, A.D.; Képíró, L.; Mockenhaupt, M.; Oon, H.H.; et al. AP1S3 Mutations Cause Skin Autoinflammation by Disrupting Keratinocyte Autophagy and Up-Regulating IL-36 Production. J. Investig. Dermatol. 2016, 136, 2251–2259. [Google Scholar] [CrossRef]
- Haskamp, S.; Bruns, H.; Hahn, M.; Hoffmann, M.; Gregor, A.; Löhr, S.; Hahn, J.; Schauer, C.; Ringer, M.; Flamann, C.; et al. Myeloperoxidase Modulates Inflammation in Generalized Pustular Psoriasis and Additional Rare Pustular Skin Diseases. Am. J. Hum. Genet. 2020, 107, 527–538. [Google Scholar] [CrossRef]
- Frey, S.; Sticht, H.; Wilsmann-Theis, D.; Gerschütz, A.; Wolf, K.; Löhr, S.; Haskamp, S.; Frey, B.; Hahn, M.; Ekici, A.B.; et al. Rare Loss-of-Function Mutation in SERPINA3 in Generalized Pustular Psoriasis. J. Investig. Dermatol. 2020, 140, 1451–1455.e1413. [Google Scholar] [CrossRef]
- Assan, F.; Husson, B.; Hegazy, S.; Seneschal, J.; Aubin, F.; Mahé, E.; Jullien, D.; Sbidian, E.; D’Incan, M.; Conrad, C.; et al. Palmoplantar pustulosis and acrodermatitis continua of Hallopeau: Demographic and clinical comparative study in a large multicentre cohort. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1578–1583. [Google Scholar] [CrossRef]
- Gisondi, P.; Bellinato, F.; Girolomoni, G. Clinical Characteristics of Patients with Pustular Psoriasis: A Single-Center Retrospective Observational Study. Vaccines 2022, 10, 1171. [Google Scholar] [CrossRef]
- Browne, S.K.; Burbelo, P.D.; Chetchotisakd, P.; Suputtamongkol, Y.; Kiertiburanakul, S.; Shaw, P.A.; Kirk, J.L.; Jutivorakool, K.; Zaman, R.; Ding, L.; et al. Adult-Onset Immunodeficiency in Thailand and Taiwan. N. Engl. J. Med. 2012, 367, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Jutivorakool, K.; Sittiwattanawong, P.; Kantikosum, K.; Hurst, C.P.; Kumtornrut, C.; Asawanonda, P.; Klaewsongkram, J.; Rerknimitr, P. Skin Manifestations in Patients with Adult-onset Immunodeficiency due to Anti-interferon-γ Autoantibody: A Relationship with Systemic Infections. Acta Derm. Venereol. 2018, 98, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Kantaputra, P.; Daroontum, T.; Chuamanochan, M.; Chaowattanapanit, S.; Kiratikanon, S.; Choonhakarn, C.; Intachai, W.; Olsen, B.; Tongsima, S.; Ngamphiw, C.; et al. SERPINB3, Adult-Onset Immunodeficiency, and Generalized Pustular Psoriasis. Genes 2023, 14, 266. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Chuamanochan, M.; Kiratikanon, S.; Chiewchanvit, S.; Chaiwarith, R.; Intachai, W.; Quarto, N.; Tongsima, S.; McGrath, J.A.; Ngamphiw, C. A truncating variant in SERPINA3, skin pustules and adult-onset immunodeficiency. J. Dermatol. 2021, 48, e370–e371. [Google Scholar] [CrossRef]
- Kantaputra, P.; Chaowattanapanit, S.; Kiratikanon, S.; Chaiwarith, R.; Choonhakarn, C.; Intachai, W.; Quarto, N.; Tongsima, S.; Ketudat Cairns, J.R.; Ngamphiw, C.; et al. SERPINA1, generalized pustular psoriasis, and adult-onset immunodeficiency. J. Dermatol. 2021, 48, 1597–1601. [Google Scholar] [CrossRef]
- Bassoy, E.Y.; Towne, J.E.; Gabay, C. Regulation and function of interleukin-36 cytokines. Immunol. Rev. 2018, 281, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Takeichi, T.; Kono, M.; Ogawa, Y.; Shimoyama, Y.; Muro, Y.; Akiyama, M. A novel IL36RN/IL1F5 homozygous nonsense mutation, p.Arg10X, in a Japanese patient with adult-onset generalized pustular psoriasis. Br. J. Dermatol. 2012, 167, 699–701. [Google Scholar] [CrossRef]
- Sugiura, K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants. J. Dermatol. Sci. 2014, 74, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Choon, S.E.; Navarini, A.A.; Pinter, A. Clinical Course and Characteristics of Generalized Pustular Psoriasis. Am. J. Clin. Dermatol. 2022, 23 (Suppl. S1), 21–29. [Google Scholar] [CrossRef]
- Körber, A.; Mössner, R.; Renner, R.; Sticht, H.; Wilsmann-Theis, D.; Schulz, P.; Sticherling, M.; Traupe, H.; Hüffmeier, U. Mutations in IL36RN in patients with generalized pustular psoriasis. J. Investig. Dermatol. 2013, 133, 2634–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Mellett, M.; Meier, B.; Mohanan, D.; Schairer, R.; Cheng, P.; Satoh, T.K.; Kiefer, B.; Ospelt, C.; Nobbe, S.; Thome, M.; et al. CARD14 Gain-of-Function Mutation Alone Is Sufficient to Drive IL-23/IL-17-Mediated Psoriasiform Skin Inflammation In Vivo. J. Investig. Dermatol. 2018, 138, 2010–2023. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Muto, M.; Akiyama, M. CARD14 c.526G>C (p.Asp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J. Investig. Dermatol. 2014, 134, 1755–1757. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; You, J.; Fu, X.; Wang, Z.; Sun, Y.; Liu, H.; Zhang, F. Variants of CARD14 are predisposing factors for generalized pustular psoriasis (GPP) with psoriasis vulgaris but not for GPP alone in a Chinese population. Br. J. Dermatol. 2019, 180, 425–426. [Google Scholar] [CrossRef]
- Ren, X.; Farías, G.G.; Canagarajah, B.J.; Bonifacino, J.S.; Hurley, J.H. Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 2013, 152, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Vergnano, M.; Mockenhaupt, M.; Benzian-Olsson, N.; Paulmann, M.; Grys, K.; Mahil, S.K.; Chaloner, C.; Barbosa, I.A.; August, S.; Burden, A.D.; et al. Loss-of-Function Myeloperoxidase Mutations Are Associated with Increased Neutrophil Counts and Pustular Skin Disease. Am. J. Hum. Genet. 2020, 107, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Onitsuka, M.; Farooq, M.; Iqbal, M.N.; Yasuno, S.; Shimomura, Y. A homozygous loss-of-function variant in the MPO gene is associated with generalized pustular psoriasis. J. Dermatol. 2022, 50, 664–671. [Google Scholar] [CrossRef]
- Vidalino, L.; Doria, A.; Quarta, S.; Zen, M.; Gatta, A.; Pontisso, P. SERPINB3, apoptosis and autoimmunity. Autoimmun. Rev. 2009, 9, 108–112. [Google Scholar] [CrossRef]
- Turato, C.; Pontisso, P. SERPINB3 (serpin peptidase inhibitor, clade B (ovalbumin), member 3). Atlas Genet. Cytogenet. Oncol. Haematol. 2015, 19, 202–209. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, P.; Wang, Z.; Sun, L.; Li, W.; Zhao, Q.; Liu, T.; Pan, Q.; Sun, Y.; Jia, F.; et al. Identification of the BTN3A3 gene as a molecule implicated in generalized pustular psoriasis in a Chinese population. J. Investig. Dermatol. 2023. [Google Scholar] [CrossRef]
- Doi, H.; Shibata, M.A.; Kiyokane, K.; Otsuki, Y. Downregulation of TGFbeta isoforms and their receptors contributes to keratinocyte hyperproliferation in psoriasis vulgaris. J. Dermatol. Sci. 2003, 33, 7–16. [Google Scholar] [CrossRef]
- Jiang, M.; Sun, Z.; Dang, E.; Li, B.; Fang, H.; Li, J.; Gao, L.; Zhang, K.; Wang, G. TGFβ/SMAD/microRNA-486-3p Signaling Axis Mediates Keratin 17 Expression and Keratinocyte Hyperproliferation in Psoriasis. J. Investig. Dermatol. 2017, 137, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Kantaputra, P.; Daroontum, T.; Chuamanochan, M.; Chaowattanapanit, S.; Intachai, W.; Olsen, B.; Sastraruji, T.; Tongsima, S.; Ngamphiw, C.; Kampuansai, J.; et al. Loss of Function TGFBR2 Variant as a Contributing Factor in Generalized Pustular Psoriasis and Adult-Onset Immunodeficiency. Genes 2022, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, S.; Zeng, J. TGF-β signaling: A complex role in tumorigenesis (Review). Mol. Med. Rep. 2018, 17, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Uppala, R.; Tsoi, L.C.; Harms, P.W.; Wang, B.; Billi, A.C.; Maverakis, E.; Michelle Kahlenberg, J.; Ward, N.L.; Gudjonsson, J.E. “Autoinflammatory psoriasis”-genetics and biology of pustular psoriasis. Cell Mol. Immunol. 2021, 18, 307–317. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Bachelez, H.; Choon, S.E.; Marrakchi, S.; Burden, A.D.; Tsai, T.F.; Morita, A.; Turki, H.; Hall, D.B.; Shear, M.; Baum, P.; et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N. Engl. J. Med. 2019, 380, 981–983. [Google Scholar] [CrossRef]
- Kanazawa, N. Designation of Autoinflammatory Skin Manifestations With Specific Genetic Backgrounds. Front. Immunol. 2020, 11, 475. [Google Scholar] [CrossRef] [Green Version]
- Müller, V.L.; Kreuter, A. Remission of recalcitrant generalized pustular psoriasis under interleukin-36 receptor inhibitor spesolimab. Dermatologie 2023, 74, 356–3594. [Google Scholar] [CrossRef]
- Ingelheim, B. European Comission Approves SPEVIGO (spesolimab) for Generalized Pustular Psoriasis Flares. Available online: https://www.boehringer-ingelheim.com/human-health/skin-diseases/gpp/european-commission-approves-spevigo-spesolimab-generalized (accessed on 14 May 2023).
- Baum, P.; Visvanathan, S.; Garcet, S.; Roy, J.; Schmid, R.; Bossert, S.; Lang, B.; Bachelez, H.; Bissonnette, R.; Thoma, C.; et al. Pustular psoriasis: Molecular pathways and effects of spesolimab in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2022, 149, 1402–1412. [Google Scholar] [CrossRef]
- Kodali, N.; Blanchard, I.; Kunamneni, S.; Lebwohl, M.G. Current management of generalized pustular psoriasis. Exp. Dermatol. 2023. [Google Scholar] [CrossRef]
- Burden, A.D.; Choon, S.E.; Gottlieb, A.B.; Navarini, A.A.; Warren, R.B. Clinical Disease Measures in Generalized Pustular Psoriasis. Am. J. Clin. Dermatol. 2022, 23 (Suppl. S1), 39–50. [Google Scholar] [CrossRef]
- Bachelez, H.; Choon, S.E.; Marrakchi, S.; Burden, A.D.; Tsai, T.F.; Morita, A.; Navarini, A.A.; Zheng, M.; Xu, J.; Turki, H.; et al. Trial of Spesolimab for Generalized Pustular Psoriasis. N. Engl. J. Med. 2021, 385, 2431–2440. [Google Scholar] [CrossRef]
- Morita, A.; Tsai, T.F.; Yee, E.Y.W.; Okubo, Y.; Imafuku, S.; Zheng, M.; Li, L.; Quaresma, M.; Thoma, C.; Choon, S.E. Efficacy and safety of spesolimab in Asian patients with a generalized pustular psoriasis flare: Results from the randomized, double-blind, placebo-controlled Effisayil™ 1 study. J. Dermatol. 2023, 50, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Elewski, B.; Lebwohl, M.G.; Anadkat, M.J.; Barker, J.; Ghoreschi, K.; Imafuku, S.; Mrowietz, U.; Li, L.; Quaresma, M.; Thoma, C.; et al. Rapid and sustained improvements in GPPGA scores with spesolimab for treatment of generalized pustular psoriasis flares in the randomized, placebo-controlled Effisayil 1 study. J. Am. Acad. Dermatol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.B.; Reich, A.; Kaszuba, A.; Placek, W.; Griffiths, C.E.M.; Zhou, J.; Randazzo, B.; Lizzul, P.; Gudjonsson, J.E. Imsidolimab, an Anti-IL-36 Receptor Monoclonal Antibody for the Treatment of Generalised Pustular Psoriasis: Results from the Phase 2 GALLOP Trial. Br. J. Dermatol. 2023, ljad083. [Google Scholar] [CrossRef] [PubMed]
- Todorović, V.; Su, Z.; Putman, C.B.; Kakavas, S.J.; Salte, K.M.; McDonald, H.A.; Wetter, J.B.; Paulsboe, S.E.; Sun, Q.; Gerstein, C.E.; et al. Small Molecule IL-36γ Antagonist as a Novel Therapeutic Approach for Plaque Psoriasis. Sci. Rep. 2019, 9, 9089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahil, S.K.; Catapano, M.; Di Meglio, P.; Dand, N.; Ahlfors, H.; Carr, I.M.; Smith, C.H.; Trembath, R.C.; Peakman, M.; Wright, J.; et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci. Transl. Med. 2017, 9, eaan2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.J.; Yu, X.; Yuan, X.R.; Chen, B.J.; Cai, N.; Zeng, S.; Sun, Y.S.; Li, H.W. The Role of IL-36 in the Pathophysiological Processes of Autoimmune Diseases. Front. Pharmacol. 2021, 12, 727956. [Google Scholar] [CrossRef]
- Zarezadeh Mehrabadi, A.; Aghamohamadi, N.; Khoshmirsafa, M.; Aghamajidi, A.; Pilehforoshha, M.; Massoumi, R.; Falak, R. The roles of interleukin-1 receptor accessory protein in certain inflammatory conditions. Immunology 2022, 166, 38–46. [Google Scholar] [CrossRef]
- Iznardo, H.; Puig, L. Exploring the Role of IL-36 Cytokines as a New Target in Psoriatic Disease. Int. J. Mol. Sci. 2021, 22, 4344. [Google Scholar] [CrossRef] [PubMed]
- Furue, K.; Yamamura, K.; Tsuji, G.; Mitoma, C.; Uchi, H.; Nakahara, T.; Kido-Nakahara, M.; Kadono, T.; Furue, M. Highlighting Interleukin-36 Signalling in Plaque Psoriasis and Pustular Psoriasis. Acta Derm. Venereol. 2018, 98, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Fujita, H.; Terui, T.; Hayama, K.; Akiyama, M.; Ikeda, S.; Mabuchi, T.; Ozawa, A.; Kanekura, T.; Kurosawa, M.; Komine, M.; et al. Japanese guidelines for the management and treatment of generalized pustular psoriasis: The new pathogenesis and treatment of GPP. J. Dermatol. 2018, 45, 1235–1270. [Google Scholar] [CrossRef]
- Kołt-Kamińska, M.; Żychowska, M.; Reich, A. Infliximab in Combination with Low-Dose Acitretin in Generalized Pustular Psoriasis: A Report of Two Cases and Review of the Literature. Biologics 2021, 15, 317–327. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, W.; Gao, Y.; Chen, F.; Yu, N.; Ding, Y.; Liu, N. Clinical analysis of generalized pustular psoriasis in Chinese patients: A retrospective study of 110 patients. J. Dermatol. 2021, 48, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Kara Polat, A.; Alpsoy, E.; Kalkan, G.; Aytekin, S.; Uçmak, D.; Yasak Güner, R.; Topkarcı, Z.; Yılmaz, O.; Emre, S.; Borlu, M.; et al. Sociodemographic, clinical, laboratory, treatment and prognostic characteristics of 156 generalized pustular psoriasis patients in Turkey: A multicentre case series. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Li, Y.H.; Liu, Z.; Zhang, X.; Jiang, Q.; Zhou, X.Y.; Su, W. Recalcitrant paradoxical pustular psoriasis induced by infliximab: Two case reports. World J. Clin. Cases 2021, 9, 3655–3661. [Google Scholar] [CrossRef]
- Imafuku, S.; Honma, M.; Okubo, Y.; Komine, M.; Ohtsuki, M.; Morita, A.; Seko, N.; Kawashima, N.; Ito, S.; Shima, T.; et al. Efficacy and safety of secukinumab in patients with generalized pustular psoriasis: A 52-week analysis from phase III open-label multicenter Japanese study. J. Dermatol. 2016, 43, 1011–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeki, H.; Nakagawa, H.; Nakajo, K.; Ishii, T.; Morisaki, Y.; Aoki, T.; Cameron, G.S.; Osuntokun, O.O. Efficacy and safety of ixekizumab treatment for Japanese patients with moderate to severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis: Results from a 52-week, open-label, phase 3 study (UNCOVER-J). J. Dermatol. 2017, 44, 355–362. [Google Scholar] [CrossRef]
- Yamasaki, K.; Nakagawa, H.; Kubo, Y.; Ootaki, K. Efficacy and safety of brodalumab in patients with generalized pustular psoriasis and psoriatic erythroderma: Results from a 52-week, open-label study. Br. J. Dermatol. 2017, 176, 741–751. [Google Scholar] [CrossRef]
- Kromer, C.; Wilsmann-Theis, D.; Gerdes, S.; Krebs, S.; Pinter, A.; Philipp, S.; Mössner, R. Changing within the same class: Efficacy of brodalumab in plaque psoriasis after treatment with an IL-17A blocker-a retrospective multicenter study. J. Dermatol. Treat. 2021, 32, 878–882. [Google Scholar] [CrossRef]
- Morita, A.; Okubo, Y.; Morisaki, Y.; Torisu-Itakura, H.; Umezawa, Y. Ixekizumab 80 mg Every 2 Weeks Treatment Beyond Week 12 for Japanese Patients with Generalized Pustular Psoriasis and Erythrodermic Psoriasis. Dermatol. Ther. 2022, 12, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Hoashi, T.; Saeki, H.; Kanda, N. A Case of Autoimmune Hepatitis/Primary Biliary Cholangitis Overlap Syndrome during Treatment with Brodalumab for Generalized Pustular Psoriasis. J. Nippon. Med. Sch. 2021, 88, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, M.; Mabuchi, T. New Treatment Addressing the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2020, 21, 7488. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, A.A.; Khatri, A.; Oberoi, R.K.; Othman, A.A. Exposure-Response Relationships for the Efficacy and Safety of Risankizumab in Japanese Subjects with Psoriasis. Clin. Pharmacokinet. 2020, 59, 575–589. [Google Scholar] [CrossRef]
- Schnabel, V.; Broekaert, S.M.C.; Schön, M.P.; Mössner, R. Clearance of annular pustular psoriasis with ustekinumab. Eur. J. Dermatol. 2017, 27, 296–297. [Google Scholar] [CrossRef]
- Langer, N.; Wilsmann-Theis, D.; Kromer, C.; Mohr, J.; Mössner, R. Successful therapy of acrodermatitis continua of Hallopeau with IL-23 blockers–two new cases. J. Der Dtsch. Dermatol. Ges. 2021, 19, 1504–1507. [Google Scholar] [CrossRef]
- Wang, W.M.; Jin, H.Z. Biologics in the treatment of pustular psoriasis. Expert. Opin. Drug Saf. 2020, 19, 969–980. [Google Scholar] [CrossRef]
- Hüffmeier, U.; Wätzold, M.; Mohr, J.; Schön, M.P.; Mössner, R. Successful therapy with anakinra in a patient with generalized pustular psoriasis carrying IL36RN mutations. Br. J. Dermatol. 2014, 170, 202–204. [Google Scholar] [CrossRef]
- Bachelez, H.; Barker, J.; Burden, A.D.; Navarini, A.A.; Krueger, J.G. Generalized pustular psoriasis is a disease distinct from psoriasis vulgaris: Evidence and expert opinion. Expert. Rev. Clin. Immunol. 2022, 18, 1033–1047. [Google Scholar] [CrossRef]
- Krueger, J.; Puig, L.; Thaçi, D. Treatment Options and Goals for Patients with Generalized Pustular Psoriasis. Am. J. Clin. Dermatol. 2022, 23, 51–64. [Google Scholar] [CrossRef] [PubMed]
- source
Generalized Pustular Psoriasis: Divergence of Innate and Adaptive Immunity
Abstract
1. Introduction
2. Gene Mutations in GPP
2.1. Mutations of IL-36 Receptor Antagonist
2.2. CARD14 Mutations/Variants
2.3. AP1S3 Mutations
2.4. TNIP1 Mutations
2.5. SERPINA3 Mutations
2.6. MPO Mutation
3. Immunopathogenesis
3.1. Autoinflammation and Autoimmunity in GPP
3.2. GPP as an Autoinflammatory Keratinization Disorder
3.3. IL-1/IL-36 Inflammatory Axis
3.4. IL-17/IL-36 Axis as a Bridge between Innate and Adaptive Immunity
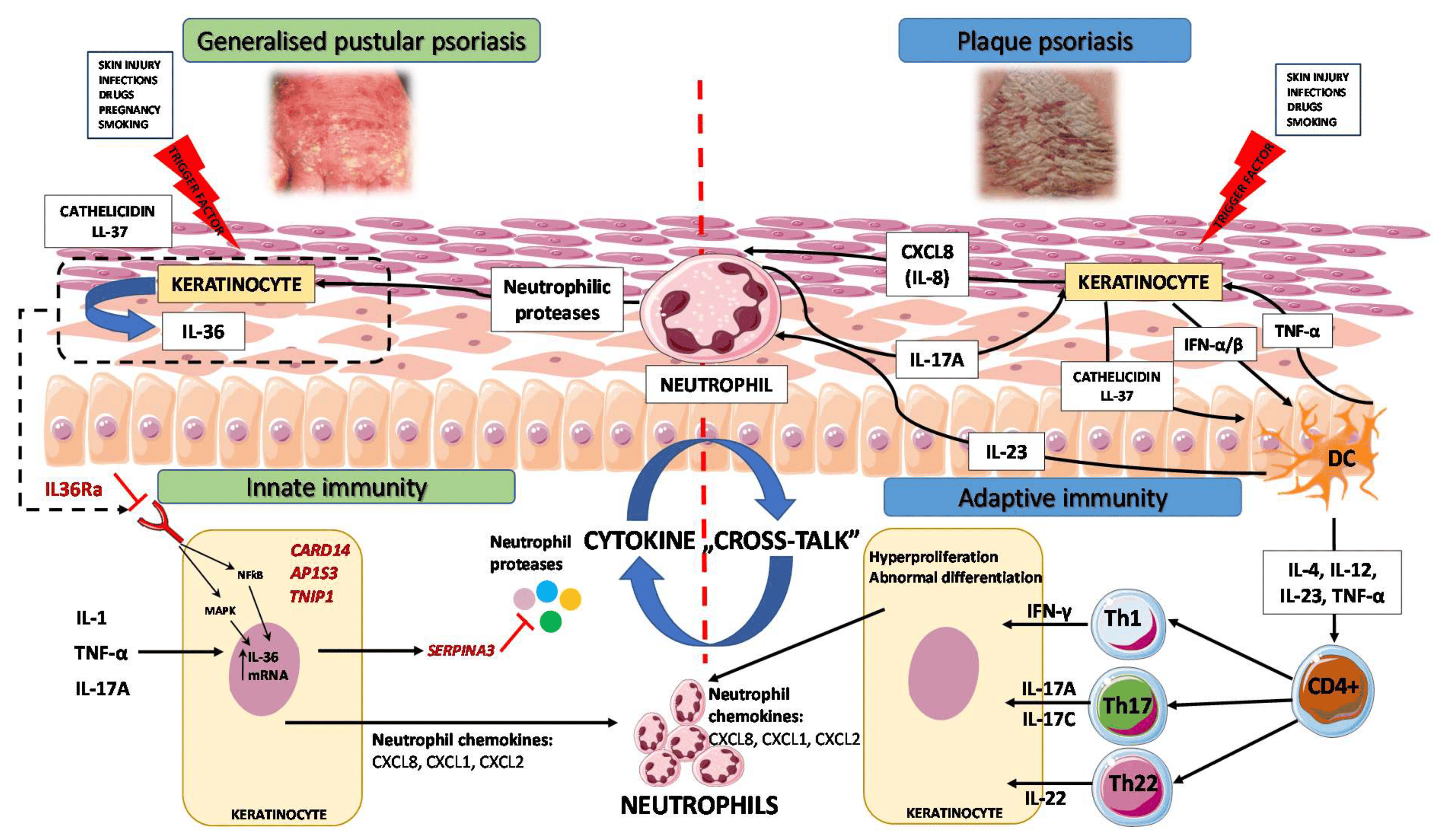
4. Biologic Therapeutics for GPP in the Light of Novel Genetic and Immunological Findings
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baker, H.; Ryan, T.J. Generalized pustular psoriasis. A clinical and epidemiological study of 104 cases. Br. J. Dermatol. 1968, 80, 771–793. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.J.; Baker, H. The prognosis of generalized pustular psoriasis. Br. J. Dermatol. 1971, 85, 407–411. [Google Scholar] [CrossRef]
- Zelickson, B.D.; Muller, S.A. Generalized pustular psoriasis. A review of 63 cases. Arch. Dermatol. 1991, 127, 1339–1345. [Google Scholar] [CrossRef] [Green Version]
- Ohkawara, A.; Yasuda, H.; Kobayashi, H.; Inaba, Y.; Ogawa, H.; Hashimoto, I.; Imamura, S. Generalized pustular psoriasis in Japan: Two distinct groups formed by differences in symptoms and genetic background. Acta Derm. Venereol. 1996, 76, 68–71. [Google Scholar]
- Augey, F.; Renaudier, P.; Nicolas, J.F. Generalized pustular psoriasis (Zumbusch): A French epidemiological survey. Eur. J. Dermatol. 2006, 16, 669–673. [Google Scholar]
- Ito, T.; Takahashi, H.; Kawada, A.; Iizuka, H.; Nakagawa, H.; Japanese Society for Psoriasis Research. Epidemiological survey from 2009 to 2012 of psoriatic patients in Japanese Society for Psoriasis Research. J. Dermatol. 2018, 45, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Nakamura, K.; Kaneko, F.; Nakagawa, H.; Iizuka, H.; Japanese Society for Psoriasis Research. Analysis of psoriasis patients registered with the Japanese Society for Psoriasis Research from 2002–2008. J. Dermatol. 2011, 38, 1125–1129. [Google Scholar] [CrossRef]
- Twelves, S.; Mostafa, A.; Dand, N.; Burri, E.; Farkas, K.; Wilson, R.; Cooper, H.L.; Irvine, A.D.; Oon, H.H.; Kingo, K.; et al. Clinical and genetic differences between pustular psoriasis subtypes. J. Allergy Clin. Immunol. 2019, 143, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Cho, H.H.; Kim, W.J.; Mun, J.H.; Song, M.; Kim, H.S.; Ko, H.C.; Kim, M.B.; Kim, H.; Kim, B.S. Clinical features and course of generalized pustular psoriasis in Korea. J. Dermatol. 2015, 42, 674–678. [Google Scholar] [CrossRef]
- Langley, R.G.; Krueger, G.G.; Griffiths, C.E. Psoriasis: Epidemiology, clinical features, and quality of life. Ann. Rheum. Dis. 2005, 64 (Suppl. 2), ii18–ii23, discussion ii24–ii25. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, C.; Barker, J. Psoriasis. In Rook’s Textbook of Dermatology, 8th ed.; Burns, T., Cox, N., Griffiths, C., Eds.; Wiley-Blackwell: Chichester, UK, 2010. [Google Scholar]
- Borges-Costa, J.; Silva, R.; Goncalves, L.; Filipe, P.; Soares de Almeida, L.; Marques Gomes, M. Clinical and laboratory features in acute generalized pustular psoriasis: A retrospective study of 34 patients. Am. J. Clin. Dermatol. 2011, 12, 271–276. [Google Scholar] [CrossRef]
- Viguier, M.; Allez, M.; Zagdanski, A.M.; Bertheau, P.; de Kerviler, E.; Rybojad, M.; Morel, P.; Dubertret, L.; Lémann, M.; Bachelez, H. High frequency of cholestasis in generalized pustular psoriasis: Evidence for neutrophilic involvement of the biliary tract. Hepatology 2004, 40, 452–458. [Google Scholar] [CrossRef]
- Bachelez, H. Pustular psoriasis and related pustular skin diseases. Br. J. Dermatol. 2018, 178, 614–618. [Google Scholar] [CrossRef]
- Choon, S.E.; Lai, N.M.; Mohammad, N.A.; Nanu, N.M.; Tey, K.E.; Chew, S.F. Clinical profile, morbidity, and outcome of adult-onset generalized pustular psoriasis: Analysis of 102 cases seen in a tertiary hospital in Johor, Malaysia. Int. J. Dermatol. 2014, 53, 676–684. [Google Scholar] [CrossRef]
- Navarini, A.A.; Burden, A.D.; Capon, F.; Mrowietz, U.; Puig, L.; Köks, S.; Kingo, K.; Smith, C.; Barker, J.N.; ERASPEN Network. European consensus statement on phenotypes of pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1792–1799. [Google Scholar] [CrossRef] [Green Version]
- Umezawa, Y.; Ozawa, A.; Kawasima, T.; Shimizu, H.; Terui, T.; Tagami, H.; Ikeda, S.; Ogawa, H.; Kawada, A.; Tezuka, T.; et al. Therapeutic guidelines for the treatment of generalized pustular psoriasis (GPP) based on a proposed classification of disease severity. Arch. Dermatol. Res. 2003, 295 (Suppl. 1), S43–S54. [Google Scholar] [CrossRef]
- Almutairi, D.; Sheasgreen, C.; Weizman, A.; Alavi, A. Generalized Pustular Psoriasis Induced by Infliximab in a Patient with Inflammatory Bowel Disease. J. Cutan. Med. Surg. 2018, 22, 507–510. [Google Scholar] [CrossRef]
- Wenk, K.S.; Claros, J.M.; Ehrlich, A. Flare of pustular psoriasis after initiating ustekinumab therapy. J. Dermatolog. Treat. 2012, 23, 212–214. [Google Scholar] [CrossRef]
- Kardaun, S.H.; Kuiper, H.; Fidler, V.; Jonkman, M.F. The histopathological spectrum of acute generalized exanthematous pustulosis (AGEP) and its differentiation from generalized pustular psoriasis. J. Cutan. Pathol. 2010, 37, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Sidoroff, A.; Dunant, A.; Viboud, C.; Halevy, S.; Bavinck, J.N.; Naldi, L.; Mockenhaupt, M.; Fagot, J.P.; Roujeau, J.C. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR). Br. J. Dermatol. 2007, 157, 989–996. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Q.; Wang, S. Genetic polymorphism of IL36RN in Han patients with generalized pustular psoriasis in Sichuan region of China: A case–control study. Medicine 2018, 97, e11741. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Takemoto, A.; Yamaguchi, M.; Takahashi, H.; Shoda, Y.; Mitsuma, T.; Tsuda, K.; Nishida, E.; Togawa, Y.; Nakajima, K.; et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J. Invest. Dermatol. 2013, 133, 2514–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, A.; Ohkido, M.; Haruki, Y.; Kobayashi, H.; Ohkawara, A.; Ohno, Y.; Inaba, Y.; Ogawa, H. Treatments of generalized pustular psoriasis: A multicenter study in Japan. J. Dermatol. 1999, 26, 141–149. [Google Scholar] [CrossRef]
- Boehner, A.; Navarini, A.A.; Eyerich, K. Generalized pustular psoriasis—A model disease for specific targeted immunotherapy, systematic review. Exp. Dermatol. 2018, 27, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Imafuku, S.; Honma, M.; Okubo, Y.; Komine, M.; Ohtsuki, M.; Morita, A.; Seko, N.; Kawashima, N.; Ito, S.; Shima, T.; et al. Efficacy and safety of secukinumab in patients with generalized pustular psoriasis: A 52-week analysis from phase III open-label multicenter Japanese study. J. Dermatol. 2016, 43, 1011–1017. [Google Scholar] [CrossRef]
- Fujita, H.; Terui, T.; Hayama, K.; Akiyama, M.; Ikeda, S.; Mabuchi, T.; Ozawa, A.; Kanekura, T.; Kurosawa, M.; Komine, M.; et al. Japanese Dermatological Association Guidelines Development Committee for the Guidelines for the Management and Treatment of Generalized Pustular Psoriasis. Japanese guidelines for the management and treatment of generalized pustular psoriasis: The new pathogenesis and treatment of GPP. J. Dermatol. 2018, 45, 1235–1270. [Google Scholar] [PubMed]
- Yamasaki, K.; Nakagawa, H.; Kubo, Y.; Ootaki, K.; Japanese Brodalumab Study Group. Efficacy and safety of brodalumab in patients with generalized pustular psoriasis and psoriatic erythroderma: Results from a 52-week, open-label study. Br. J. Dermatol. 2017, 176, 741–751. [Google Scholar] [CrossRef]
- Sano, S.; Kubo, H.; Morishima, H.; Goto, R.; Zheng, R.; Nakagawa, H. Guselkumab, a human interleukin-23 monoclonal antibody in Japanese patients with generalized pustular psoriasis and erythrodermic psoriasis: Efficacy and safety analyses of a 52-week, phase 3, multicenter, open-label study. J. Dermatol. 2018, 45, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.M.; Jin, H.Z. Biologics in the treatment of pustular psoriasis. Expert Opin. Drug Saf. 2020, 19, 969–980. [Google Scholar] [CrossRef]
- Zhou, J.; Luo, Q.; Cheng, Y.; Wen, X.; Liu, J. An update on genetic basis of generalized pustular psoriasis (Review). Int. J. Mol. Med. 2021, 47, 118. [Google Scholar] [CrossRef] [PubMed]
- Plachouri, K.M.; Chourdakis, V.; Georgiou, S. The role of IL-17 and IL-17 receptor inhibitors in the management of generalized pustular psoriasis. Drugs Today 2019, 55, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Gooderham, M.J.; Van Voorhees, A.S.; Lebwohl, M.G. An update on generalized pustular psoriasis. Expert Rev. Clin. Immunol. 2019, 15, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbidian, E.; Chaimani, A.; Afach, S.; Doney, L.; Dressler, C.; Hua, C.; Mazaud, C.; Phan, C.; Hughes, C.; Riddle, D.; et al. Systemic pharmacological treatments for chronic plaque psoriasis: A network meta-analysis. Cochrane Database Syst. Rev. 2020, 9, 1, CD011535. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Van Voorhees, A.S.; Hsu, S.; Korman, N.J.; Lebwohl, M.G.; Bebo, B.F., Jr.; Kalb, R.E. Treatment of pustular psoriasis: From the Medical Board of the National Psoriasis Foundation. J. Am. Acad. Dermatol. 2012, 67, 279–288. [Google Scholar] [CrossRef]
- Collamer, A.N.; Battafarano, D.F. Psoriatic skin lesions induced by tumor necrosis factor antagonist therapy: Clinical features and possible immunopathogenesis. Semin. Arthritis Rheum. 2010, 40, 233–240. [Google Scholar] [CrossRef]
- Kucharekova, M.; Winnepenninckx, V.; Frank, J.; Poblete-Gutiérrez, P. Generalized pustulosis induced by adalimumab in a patient with rheumatoid arthritis—A therapeutic challenge. Int. J. Dermatol. 2008, 47 (Suppl. 1), 25–28. [Google Scholar] [CrossRef]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A mixed autoimmune and autoinflammatory disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef]
- Liang, Y.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.K.; Roberts, L.W.; Voorhees, J.J.; Kahlenberg, J.M.; Harms, P.W.; Johnston, A.; et al. Six-transmembrane epithelial antigens of the prostate comprise a novel inflammatory nexus in patients with pustular skin disorders. J. Allergy Clin. Immunol. 2017, 139, 1217–1227. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Masters, S.L.; Ferguson, P.J.; Dancey, P.; Frenkel, J.; van Royen-Kerkhoff, A.; Laxer, R.; Tedgård, U.; Cowen, E.W.; Pham, T.H.; et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 2009, 360, 2426–2437. [Google Scholar] [CrossRef] [Green Version]
- Jesus, A.A.; Osman, M.; Silva, C.A.; Kim, P.W.; Pham, T.H.; Gadina, M.; Yang, B.; Bertola, D.R.; Carneiro-Sampaio, M.; Ferguson, P.J.; et al. A novel mutation of IL1RN in the deficiency of interleukin-1 receptor antagonist syndrome: Description of two unrelated cases from Brazil. Arthritis Rheum. 2011, 63, 4007–4017. [Google Scholar] [CrossRef] [Green Version]
- Minkis, K.; Aksentijevich, I.; Goldbach-Mansky, R.; Magro, C.; Scott, R.; Davis, J.G.; Sardana, N.; Herzog, R. Interleukin 1 receptor antagonist deficiency presenting as infantile pustulosis mimicking infantile pustular psoriasis. Arch. Dermatol. 2012, 148, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Jia, S.; Geoffrey, R.; Lorier, R.; Suchi, M.; Broeckel, U.; Hessner, M.J.; Verbsky, J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 2009, 360, 2438–2444. [Google Scholar] [CrossRef] [Green Version]
- Schnellbacher, C.; Ciocca, G.; Menendez, R.; Aksentijevich, I.; Goldbach-Mansky, R.; Duarte, A.M.; Rivas-Chacon, R. Deficiency of interleukin-1 receptor antagonist responsive to anakinra. Pediatr. Dermatol. 2013, 30, 758–760. [Google Scholar] [CrossRef] [Green Version]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Simpson, M.A.; Pink, A.E.; Di Meglio, P.; Smith, C.H.; Pullabhatla, V.; Knight, J.; Spain, S.L.; Nestle, F.O.; Burden, A.D.; et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011, 89, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, H.; Dinh, H.; Trueblood, E.S.; Pretorius, J.; Kugler, D.; Weng, N.; Kanaly, S.T.; Towne, J.E.; Willis, C.R.; Kuechle, M.K.; et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 2007, 204, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrowietz, U.; Burden, A.D.; Pinter, A.; Reich, K.; Schäkel, K.; Baum, P.; Datsenko, Y.; Deng, H.; Padula, S.J.; Thoma, C.; et al. Spesolimab, an Anti-Interleukin-36 Receptor Antibody, in Patients with Palmoplantar Pustulosis: Results of a Phase IIa, Multicenter, Double-Blind, Randomized, Placebo-Controlled Pilot Study. Dermatol. Ther. 2021, 11, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Setta-Kaffetzi, N.; Simpson, M.A.; Navarini, A.A.; Patel, V.M.; Lu, H.C.; Allen, M.H.; Duckworth, M.; Bachelez, H.; Burden, A.D.; Choon, S.E.; et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 2014, 94, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahil, S.K.; Twelves, S.; Farkas, K.; Setta-Kaffetzi, N.; Burden, A.D.; Gach, J.E.; Irvine, A.D.; Képíró, L.; Mockenhaupt, M.; Oon, H.H.; et al. AP1S3 Mutations Cause Skin Autoinflammation by Disrupting Keratinocyte Autophagy and Up-Regulating IL-36 Production. J. Investig. Dermatol. 2016, 136, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Berki, D.M.; Liu, L.; Choon, S.E.; David Burden, A.; Griffiths, C.E.M.; Navarini, A.A.; Tan, E.S.; Irvine, A.D.; Ranki, A.; Ogo, T.; et al. Activating CARD14 Mutations Are Associated with Generalized Pustular Psoriasis but Rarely Account for Familial Recurrence in Psoriasis Vulgaris. J. Investig. Dermatol. 2015, 135, 2964–2970. [Google Scholar] [CrossRef] [Green Version]
- Mössner, R.; Wilsmann-Theis, D.; Oji, V.; Gkogkolou, P.; Löhr, S.; Schulz, P.; Körber, A.; Prinz, J.C.; Renner, R.; Schäkel, K.; et al. The genetic basis for most patients with pustular skin disease remains elusive. Br. J. Dermatol. 2018, 178, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef] [Green Version]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Bassoy, E.Y.; Towne, J.E.; Gabay, C. Regulation and function of interleukin-36 cytokines. Immunol. Rev. 2018, 281, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Capon, F. IL36RN mutations in generalized pustular psoriasis: Just the tip of the iceberg? J. Investig. Dermatol. 2013, 133, 2503–2504. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.; Nakai, H.; Fujimoto, A.; Fujikawa, H.; Matsuyama, A.; Kariya, N.; Aizawa, A.; Fujiwara, H.; Ito, M.; Shimomura, Y. Mutation analysis of the IL36RN gene in 14 Japanese patients with generalized pustular psoriasis. Hum. Mutat. 2013, 34, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases. J. Allergy Clin. Immunol. 2017, 140, 1545–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases: An emerging concept encompassing various inflammatory keratinization disorders of the skin. J. Dermatol. Sci. 2018, 90, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, M. Autoinflammatory Keratinization Diseases (AiKDs): Expansion of Disorders to Be Included. Front. Immunol. 2020, 11, 280. [Google Scholar] [CrossRef] [Green Version]
- Uppala, R.; Tsoi, L.C.; Harms, P.W.; Wang, B.; Bill, A.C.; Maverakis, E.; Kahlenberg, M.J.; Ward, N.L.; Gudjonsson, J.E. “Autoinflammatory psoriasis”—genetics and biology of pustular psoriasis. Cell. Mol. Immunol. 2021, 18, 307–317. [Google Scholar] [CrossRef]
- Hussain, S.; Berki, D.M.; Choon, S.E.; Burden, A.D.; Allen, M.H.; Arostegui, J.I.; Chaves, A.; Duckworth, M.; Irvine, A.D.; Mockenhaupt, M.; et al. IL36RN mutations define a severe autoinflammatory phenotype of generalized pustular psoriasis. J. Allergy Clin. Immunol. 2015, 135, 1067–1070.e9. [Google Scholar] [CrossRef]
- Wang, T.S.; Chiu, H.Y.; Hong, J.B.; Chan, C.C.; Lin, S.J.; Tsai, T.F. Correlation of IL36RN mutation with different clinical features of pustular psoriasis in Chinese patients. Arch. Dermatol. Res. 2016, 308, 55–63. [Google Scholar] [CrossRef]
- Bachelez, H. Pustular Psoriasis: The Dawn of a New Era. Acta Derm. Venereol. 2020, 100, adv00034. [Google Scholar] [CrossRef] [Green Version]
- Fuchs-Telem, D.; Sarig, O.; van Steensel, M.A.; Isakov, O.; Israeli, S.; Nousbeck, J.; Richard, K.; Winnepenninckx, V.; Vernooij, M.; Shomron, N.; et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am. J. Hum. Genet. 2012, 91, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Blonska, M.; Lin, X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol. Rev. 2009, 228, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeichi, T.; Akiyama, M. Generalized Pustular Psoriasis: Clinical Management and Update on Autoinflammatory Aspects. Am. J. Clin. Dermatol. 2020, 21, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Fang, H.; Zhang, J.; Jiang, M.; Xue, K.; Ma, J.; Zhang, J.; Lei, J.; Zhang, Y.; Li, B.; et al. Neutrophil exosomes enhance the skin autoinflammation in generalized pustular psoriasis via activating keratinocytes. FASEB J. 2019, 33, 6813–6828. [Google Scholar] [CrossRef]
- Sugiura, K.; Muto, M.; Akiyama, M. CARD14 c.526G > C (pAsp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J. Investig. Dermatol. 2014, 134, 1755–1757. [Google Scholar] [CrossRef] [Green Version]
- Takeichi, T.; Sugiura, K.; Nomura, T.; Sakamoto, T.; Ogawa, Y.; Oiso, N.; Futei, Y.; Fujisaki, A.; Koizumi, A.; Aoyama, Y.; et al. Pityriasis Rubra Pilaris Type V as an Autoinflammatory Disease by CARD14 Mutations. JAMA Dermatol. 2017, 153, 66–70. [Google Scholar] [CrossRef]
- Heyninck, K.; Kreike, M.M.; Beyaert, R. Structure-function analysis of the A20-binding inhibitor of NF-kappa B activation, ABIN-1. FEBS Lett. 2003, 536, 135–140. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Y.; Zhang, Z.; Lin, J.; Chen, G.; Han, L.; Fang, X.U.; Huang, Q.; Xu, J. Identification of Two Loci Associated with Generalized Pustular Psoriasis. J. Investig. Dermatol. 2015, 135, 2132–2134. [Google Scholar] [CrossRef] [Green Version]
- Han, J.W.; Wang, Y.; Alateng, C.; Li, H.B.; Bai, Y.H.; Lyu, X.X.; Wu, R. Tumor Necrosis Factor-alpha Induced Protein 3 Interacting Protein 1 Gene Polymorphisms and Pustular Psoriasis in Chinese Han Population. Chin. Med. J. 2016, 129, 1519–1524. [Google Scholar] [CrossRef]
- Cooperman, B.S.; Stavridi, E.; Nickbarg, E.; Rescorla, E.; Schechter, N.M.; Rubin, H. Antichymotrypsin interaction with chymotrypsin. Partitioning of the complex. J. Biol. Chem. 1993, 268, 23616–23625. [Google Scholar] [CrossRef]
- Frey, S.; Sticht, H.; Wilsmann-Theis, D.; Gerschütz, A.; Wolf, K.; Löhr, S.; Haskamp, S.; Frey, B.; Hahn, M.; Ekici, A.B.; et al. Rare Loss-of-Function Mutation in SERPINA3 in Generalized Pustular Psoriasis. J. Investig. Dermatol. 2020, 140, 1451–1455.e13. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tu, J.; Hu, Y.; Song, G.; Yin, Z. Cathepsin G cleaves and activates IL-36γ and promotes the inflammation of psoriasis. Drug Des. Devel. Ther. 2019, 13, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Austin, G.E.; Chan, W.C.; Zhao, W.; Racine, M. Myeloperoxidase gene expression in normal granulopoiesis and acute leukemias. Leuk. Lymphoma. 1994, 15, 209–226. [Google Scholar] [CrossRef]
- De Argila, D.; Dominguez, J.D.; Lopez-Estebaranz, J.L.; Iglesias, L. Pustular psoriasis in a patient with myeloperoxidase deficiency. Dermatology 1996, 193, 270. [Google Scholar] [CrossRef]
- Vergnano, M.; Mockenhaupt, M.; Benzian-Olsson, N.; Paulmann, M.; Grys, K.; Mahil, S.K.; Chaloner, C.; Barbosa, I.A.; August, S.; Burden, A.D.; et al. Loss-of-Function Myeloperoxidase Mutations Are Associated with Increased Neutrophil Counts and Pustular Skin Disease. Am. J. Hum. Genet. 2020, 107, 539–543. [Google Scholar] [CrossRef]
- Kizaki, M.; Miller, C.W.; Selsted, M.E.; Koeffler, H.P. Myeloperoxidase (MPO) gene mutation in hereditary MPO deficiency. Blood 1994, 83, 1935–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, C.; Patriarca, P.; Solero, G.P.; Baralle, F.E.; Romano, M. Genetic studies on myeloperoxidase deficiency in Italy. Jpn. J. Infect. Dis. 2004, 57, S10–S12. [Google Scholar]
- Haskamp, S.; Bruns, H.; Hahn, M.; Hoffmann, M.; Gregor, A.; Löhr, S.; Hahn, J.; Schauer, C.; Ringer, M.; Flamann, C.; et al. Myeloperoxidase Modulates Inflammation in Generalized Pustular Psoriasis and Additional Rare Pustular Skin Diseases. Am. J. Hum. Genet. 2020, 107, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Viguier, M.; Guigue, P.; Pagès, C.; Smahi, A.; Bachelez, H. Successful treatment of generalized pustular psoriasis with the interleukin-1-receptor antagonist Anakinra: Lack of correlation with IL1RN mutations. Ann. Intern. Med. 2010, 153, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Hüffmeier, U.; Wätzold, M.; Mohr, J.; Schön, M.P.; Mössner, R. Successful therapy with anakinra in a patient with generalized pustular psoriasis carrying IL36RN mutations. Br. J. Dermatol. 2014, 170, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Papagoras, C.; Lefaki, I.; Giatromanolaki, A.; Kotsianidis, I.; Speletas, M.; Bocly, V.; Theodorou, I.; Dalla, V.; Ritis, K. Successful response in a case of severe pustular psoriasis after interleukin-1β inhibition. Br. J. Dermatol. 2017, 176, 212–215. [Google Scholar] [CrossRef]
- Mansouri, B.; Richards, L.; Menter, A. Treatment of two patients with generalized pustular psoriasis with the interleukin-1β inhibitor gevokizumab. Br. J. Dermatol. 2015, 173, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; Choon, S.E.; Marrakchi, S.; Burden, A.D.; Tsai, T.F.; Morita, A.; Turki, H.; Hall, D.B.; Shear, M.; Baum, P.; et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N. Engl. J. Med. 2019, 380, 981–983. [Google Scholar] [CrossRef]
- Choon, S.E.; Lebwohl, M.G.; Marrakchi, S.; Burden, A.D.; Tsai, T.F.; Morita, A.; Navarini, A.A.; Zheng, M.; Xu, J.; Turki, H.; et al. Study protocol of the global Effisayil 1 Phase II, multicentre, randomised, double-blind, placebo-controlled trial of spesolimab in patients with generalized pustular psoriasis presenting with an acute flare. BMJ Open 2021, 11, e043666. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A 5-year Study to Test BI 655130 in Patients with Generalized Pustular Psoriasis Who Took Part in Previous Studies with BI 655130. NCT03886246. Available online: https://clinicaltrials.gov/ct2/show/NCT03886246 (accessed on 15 May 2021).
- ClinicalTrials.gov. A Study to Evaluate the Efficacy and Safety of ANB019 in Subjects with Generalized Pustular Psoriasis (GPP). NCT03619902. Available online: https://clinicaltrials.gov/ct2/show/NCT03619902 (accessed on 15 May 2021).
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Brydges, S.; Kastner, D.L. The systemic autoinflammatory diseases: Inborn errors of the innate immune system. Curr. Top. Microbiol. Immunol. 2006, 305, 127–160. [Google Scholar] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Ma, H.L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, C.; Towne, J.E. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J. Leukoc. Biol. 2015, 97, 645–652. [Google Scholar] [CrossRef]
- Mudigonda, P.; Mudigonda, T.; Feneran, A.N.; Alamdari, H.S.; Sandoval, L.; Feldman, S.R. Interleukin-23 and interleukin-17: Importance in pathogenesis and therapy of psoriasis. Dermatol. Online J. 2012, 18, 1. [Google Scholar] [CrossRef]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef]
- Goldstein, J.D.; Bassoy, E.Y.; Caruso, A.; Palomo, J.; Rodriguez, E.; Lemeille, S.; Gabay, C. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci. Alliance 2020, 3, e202000688. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, A.; Vollmer, S.; Besgen, P.; Galinski, A.; Summer, B.; Kawakami, Y.; Wollenberg, A.; Dornmair, K.; Spannagl, M.; Ruzicka, T.; et al. Unopposed IL-36 Activity Promotes Clonal CD4+ T-Cell Responses with IL-17A Production in Generalized Pustular Psoriasis. J. Investig. Dermatol. 2018, 138, 1338–1347. [Google Scholar] [CrossRef] [Green Version]
- Trent, J.T.; Kerdel, F.A. Successful treatment of Von Zumbusch pustular psoriasis with infliximab. J. Cutan. Med. Surg. 2004, 8, 224–228. [Google Scholar] [CrossRef]
- Martin, D.A.; Towne, J.E.; Kricorian, G.; Klekotka, P.; Gudjonsson, J.E.; Krueger, J.G.; Russell, C.B. The emerging role of IL-17 in the pathogenesis of psoriasis: Preclinical and clinical findings. J. Investig. Dermatol. 2013, 133, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. 2009, 160, 319–324. [Google Scholar] [CrossRef]
- Ishigame, H.; Kakuta, S.; Nagai, T.; Kadoki, M.; Nambu, A.; Komiyama, Y.; Fujikado, N.; Tanahashi, Y.; Akitsu, A.; Kotaki, H.; et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009, 30, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Furue, K.; Yamamura, K.; Tsuji, G.; Mitoma, C.; Uchi, H.; Nakahara, T.; Kido-Nakahara, M.; Kadono, T.; Furue, M. Highlighting Interleukin-36 Signalling in Plaque Psoriasis and Pustular Psoriasis. Acta Derm. Venereol. 2018, 98, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Neuhauser, R.; Eyerich, K.; Boehner, A. Generalized pustular psoriasis-Dawn of a new era in targeted immunotherapy. Exp. Dermatol. 2020, 29, 1088–1096. [Google Scholar] [CrossRef]
- Croxford, A.L.; Karbach, S.; Kurschus, F.C.; Wörtge, S.; Nikolaev, A.; Yogev, N.; Klebow, S.; Schüler, R.; Reissig, S.; Piotrowski, C.; et al. IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J. Investig. Dermatol. 2014, 134, 728–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saggini, A.; Chimenti, S.; Chiricozzi, A. IL-6 as a druggable target in psoriasis: Focus on pustular variants. J. Immunol. Res. 2014, 2014, 964069. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.; Kotowsky, N.; Medeiros, R.; Mackey, R.H.; Harrold, L.R.; Valdecantos, W.C.; Flack, M.; Golembesky, A.K.; Lebwohl, M. Unmet Medical Needs in the Treatment and Management of Generalized Pustular Psoriasis Flares: Evidence from a Survey of Corrona Registry Dermatologists. Dermatol. Ther. 2021, 11, 529–541. [Google Scholar] [CrossRef]
- Ettehadi, P.; Greaves, M.W.; Wallach, D.; Aderka, D.; Camp, R.D. Elevated tumour necrosis factor-alpha (TNF-alpha) biological activity in psoriatic skin lesions. Clin. Exp. Immunol. 1994, 96, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qiu, L.; Xiao, T.; Chen, H.D. Juvenile generalized pustular psoriasis with IL36RN mutation treated with short-term infliximab. Dermatol. Ther. 2016, 29, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Peng, C.; Ding, Y.; Yi, X.; Gao, Y. Development of herpes zoster during infliximab treatment for pediatric generalized pustular psoriasis: A case report. Dermatol. Ther. 2019, 32, e12838. [Google Scholar] [CrossRef]
- Skrabl-Baumgartner, A.; Weger, W.; Salmhofer, W.; Jahnel, J. Childhood generalized pustular psoriasis: Longtime remission with combined infliximab and methotrexate treatment. Pediatr. Dermatol. 2015, 32, e13–e14. [Google Scholar]
- Tsang, V.; Dvorakova, V.; Enright, F.; Murphy, M.; Gleeson, C. Successful use of infliximab as first line treatment for severe childhood generalized pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, e117–e119. [Google Scholar] [CrossRef]
- Viguier, M.; Aubin, F.; Delaporte, E.; Pagès, C.; Paul, C.; Beylot-Barry, M.; Goujon, C.; Rybojad, M.; Bachelez, H.; Groupe de Recherche sur le Psoriasis de la Société Française de Dermatologie. Efficacy and safety of tumor necrosis factor inhibitors in acute generalized pustular psoriasis. Arch. Dermatol. 2012, 148, 1423–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulalhon, N.; Begon, E.; Lebbé, C.; Lioté, F.; Lahfa, M.; Bengoufa, D.; Morel, P.; Dubertret, L.; Bachelez, H. A follow-up study in 28 patients treated with infliximab for severe recalcitrant psoriasis: Evidence for efficacy and high incidence of biological autoimmunity. Br. J. Dermatol. 2007, 156, 329–336. [Google Scholar] [CrossRef]
- Matsumoto, A.; Komine, M.; Karakawa, M.; Kishimoto, M.; Ohtsuki, M. Adalimumab administration after infliximab therapy is a successful treatment strategy for generalized pustular psoriasis. J. Dermatol. 2017, 44, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Silfvast-Kaiser, A.; Paek, S.Y.; Menter, A. Anti-IL17 therapies for psoriasis. Expert Opin. Biol. Ther. 2019, 19, 45–54. [Google Scholar] [CrossRef]
- Daudén, E.; Santiago-et-Sánchez-Mateos, D.; Sotomayor-López, E.; García-Díez, A. Ustekinumab: Effective in a patient with severe recalcitrant generalized pustular psoriasis. Br. J. Dermatol. 2010, 163, 1346–1347. [Google Scholar] [CrossRef]
- Arakawa, A.; Ruzicka, T.; Prinz, J.C. Therapeutic Efficacy of Interleukin 12/Interleukin 23 Blockade in Generalized Pustular Psoriasis Regardless of IL36RN Mutation Status. JAMA Dermatol. 2016, 152, 825–828. [Google Scholar] [CrossRef]
- Storan, E.R.; O’Gorman, S.M.; Markham, T. Generalized pustular psoriasis treated with ustekinumab. Clin. Exp. Dermatol. 2016, 41, 689–690. [Google Scholar] [CrossRef]
- Markham, A. Guselkumab: First Global Approval. Drugs 2017, 77, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K.; Duggan, S. Risankizumab: First Global Approval. Drugs 2019, 79, 893–900. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A Study to Assess Efficacy and Safety of Two Different Dose Regimens of Risankizumab Administered Subcutaneously in Japanese Subjects with Generalized Pustular Psoriasis or Erythrodermic Psoriasis. NCT03022045. Available online: https://clinicaltrials.gov/ct2/show/NCT03022045 (accessed on 12 May 2021).
- Rossi-Semerano, L.; Piram, M.; Chiaverini, C.; De Ricaud, D.; Smahi, A.; Koné-Paut, I. First clinical description of an infant with interleukin-36-receptor antagonist deficiency successfully treated with anakinra. Pediatrics 2013, 132, e1043–e1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiler, J.; McDermott, M.F. Gevokizumab, an anti-IL-1β mAb for the potential treatment of type 1 and 2 diabetes, rheumatoid arthritis and cardiovascular disease. Curr. Opin. Mol. Ther. 2010, 12, 755–769. [Google Scholar]
- Ratnarajah, K.; Jfri, A.; Litvinov, I.V.; Netchiporouk, E. Spesolimab: A Novel Treatment for Pustular Psoriasis. J. Cutan. Med. Surg. 2020, 24, 199–200. [Google Scholar] [CrossRef]
- AnaptysBio Reports Positive Topline Data from GALLOP Phase 2 Clinical Trial of Imsidolimab in Moderate-to-Severe Generalized Pustular Psoriasis (GPP). AnaptysBio. News Release. 13 October 2020. Available online: https://ir.anaptysbio.com/news-releases/news-release-details/anaptysbio-reports-positive-topline-data-gallop-phase-2-clinical (accessed on 8 April 2021).
- Morita, A.; Yamazaki, F.; Matsuyama, T.; Takahashi, K.; Arai, S.; Asahina, A.; Imafuku, S.; Nakagawa, H.; Hasegawa, Y.; Williams, D.; et al. Adalimumab treatment in Japanese patients with generalized pustular psoriasis: Results of an open-label phase 3 study. J. Dermatol. 2018, 45, 1371–1380. [Google Scholar] [CrossRef] [Green Version]
- Hansel, K.; Marietti, R.; Tramontana, M.; Bianchi, L.; Romita, P.; Giuffrida, R.; Stingeni, L. Childhood generalized pustular psoriasis: Successful long-term treatment with adalimumab. Dermatol. Ther. 2020, 33, e13294. [Google Scholar] [CrossRef]
- Ho, P.H.; Tsai, T.F. Successful treatment of refractory juvenile generalized pustular psoriasis with secukinumab monotherapy: A case report and review of published work. J. Dermatol. 2018, 45, 1353–1356. [Google Scholar] [CrossRef]
- Mizutani, Y.; Mizutani, Y.H.; Matsuyama, K.; Kawamura, M.; Fujii, A.; Shu, E.; Ohnishi, H.; Seishima, M. Generalized pustular psoriasis in pregnancy, successfully treated with certolizumab pegol. J. Dermatol. 2020, 47, e262–e263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Georgakopoulos, J.R.; Ighani, A.; Yeung, J. Systemic Monotherapy Treatments for Generalized Pustular Psoriasis: A Systematic Review. J. Cutan. Med. Surg. 2018, 22, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Kromer, C.; Loewe, E.; Schaarschmidt, M.L.; Pinter, A.; Gerdes, S.; Herr, R.; Poortinga, S.; Moessner, R.; Wilsmann-Theis, D. Drug survival in the treatment of generalized pustular psoriasis: A retrospective multicenter study. Dermatol. Ther. 2021, 34, e14814. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Nakagawa, H.; Nakajo, K.; Ishii, T.; Morisaki, Y.; Aoki, T.; Cameron, G.S.; Osuntokun, O.O.; Japanese Ixekizumab Study Group. Efficacy and safety of ixekizumab treatment for Japanese patients with moderate to severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis: Results from a 52-week, open-label, phase 3 study (UNCOVER-J). J. Dermatol. 2017, 44, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.M.; Spanou, Z.; Tang, H.; Schibler, F.; Pelivani, N.; Yawalkar, N. Rapid downregulation of innate immune cells, interleukin-12 and interleukin-23 in generalized pustular psoriasis with infliximab in combination with acitretin. Dermatology 2012, 225, 338–343. [Google Scholar] [CrossRef] [PubMed]
- De Rie, M.A.; Zonneveld, I.M.; Witkamp, L. Soluble interleukin-2 receptor (sIL-2R) is a marker of disease activity in psoriasis: A comparison of sIL-2R, sCD27, sCD4, sCD8 and sICAM-1. Acta Dermatol. Venereol. 1996, 76, 357–360. [Google Scholar]
- Salim, A.; Emerson, R.M.; Dalziel, K.L. Successful treatment of severe generalized pustular psoriasis with basiliximab (interleukin-2 receptor blocker). Br. J. Dermatol. 2000, 143, 1121–1122. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study to Test Whether BI 655130 (Spesolimab) Prevents Flare-Ups in Patients with Generalized Pustular Psoriasis. NCT04399837. Available online: https://clinicaltrials.gov/ct2/show/NCT04399837 (accessed on 16 May 2021).
- source